Adverse Event Calculator: IR vs EAIR
Input Data
What You Need to Know
Understanding these metrics is crucial for evaluating drug safety:
- IR (Incidence Rate) Percentage of patients who experienced the event, regardless of treatment duration
- EAIR (Exposure-Adjusted Incidence Rate) Events per 100 patient-years, accounting for varying treatment lengths
- Patient-Year One person exposed for one year (365.25 days)
Results
Incidence Rate (IR)
Exposure-Adjusted Incidence Rate (EAIR)
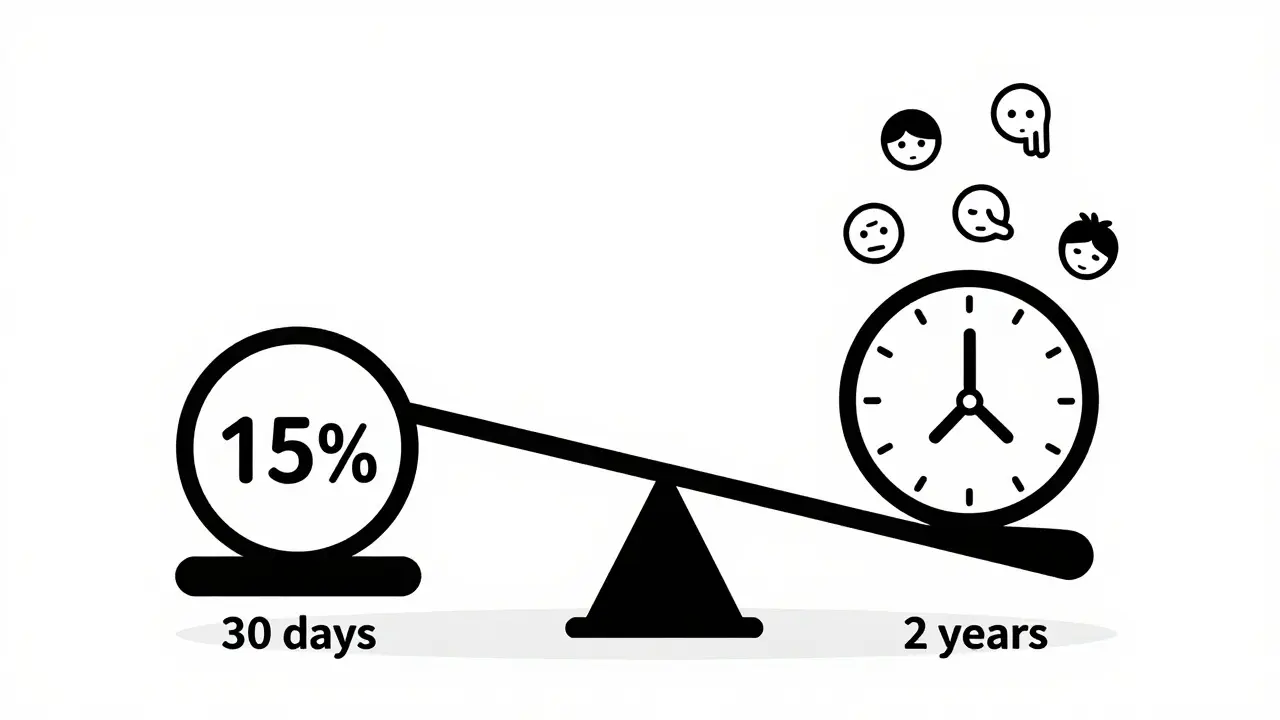
When a new drug enters clinical trials, regulators and doctors don’t just look at how well it works-they also need to know how safe it is. But here’s the problem: adverse event rates aren’t as simple as saying "15% of patients had nausea." If one group took the drug for 3 months and another for 2 years, comparing raw percentages can mislead you. That’s why experts now rely on more precise methods like exposure-adjusted incidence rate (EAIR) to get the real picture.
Why Raw Percentages Lie About Risk
The simplest way to report adverse events is called incidence rate (IR). It’s just: number of people who had the side effect divided by total people in the study. So if 15 out of 100 patients got a headache, you say "15% experienced headache." But this ignores something critical: time. Imagine two groups in a trial. Group A takes the drug for 30 days. Group B takes it for 730 days-two full years. If 10 people in Group A get a rash, that’s 10%. In Group B, 20 people get a rash. At first glance, it looks worse. But if you think about it, Group B had way more time to develop side effects. The raw percentage doesn’t tell you if the drug is actually riskier, or if people just had more time to react.A 2010 analysis cited by Amgen statisticians found that using IR alone can underestimate true event rates by 18% to 37% when treatment lengths vary. That’s not a small error-it’s enough to make a drug look safer than it really is, or worse, to wrongly scare people away from a beneficial treatment.
Enter Patient-Years: The EIR Method
To fix this, researchers use event incidence rate adjusted by patient-years (EIR). Instead of counting people, you count time. One patient-year means one person was exposed to the drug for one full year. If five people took the drug for 6 months each, that’s 2.5 patient-years.The formula is simple: number of events divided by total patient-years. Then you scale it to "per 100 patient-years" to make it easier to read. So if 15 events happened over 200 patient-years, the EIR is 7.5 per 100 patient-years.
This method works better for recurring events. Say a patient gets diarrhea twice in 18 months. IR counts them as one person with diarrhea. EIR counts two events over 1.5 patient-years-giving you a clearer sense of how often the problem actually happens. JMP Clinical and other regulatory software now use EIR as a standard output, pulling dates from trial records (TRTSDTM and TRTEDTM) to calculate exposure automatically.
The FDA’s Push for EAIR: What Changed in 2023
In 2023, the FDA made a major move. They requested that a company submitting a supplemental biologics license application (sBLA) switch from IR to exposure-adjusted incidence rate (EAIR). This wasn’t a suggestion-it was a requirement. And it signaled a turning point.EAIR doesn’t just count events per time. It accounts for recurrence and variable exposure in a way EIR doesn’t. For example, if a patient stops the drug for 3 weeks due to side effects, then restarts, EAIR adjusts for that gap. EIR might still count them as continuously exposed. EAIR doesn’t.
MSD’s safety team found that switching to EAIR revealed previously hidden safety signals in 12% of their chronic therapy programs. One drug looked fine with IR. But when they used EAIR, they noticed a spike in liver enzyme elevations in patients who took the drug for over 18 months-something IR had buried.
The European Medicines Agency (EMA) still allows both IR and EAIR, but requires justification. The FDA, however, is moving toward EAIR as the new baseline. Dr. Gary Koch, former biostatistics professor at UNC, called ignoring exposure time a "fundamental statistical error." He wasn’t exaggerating.
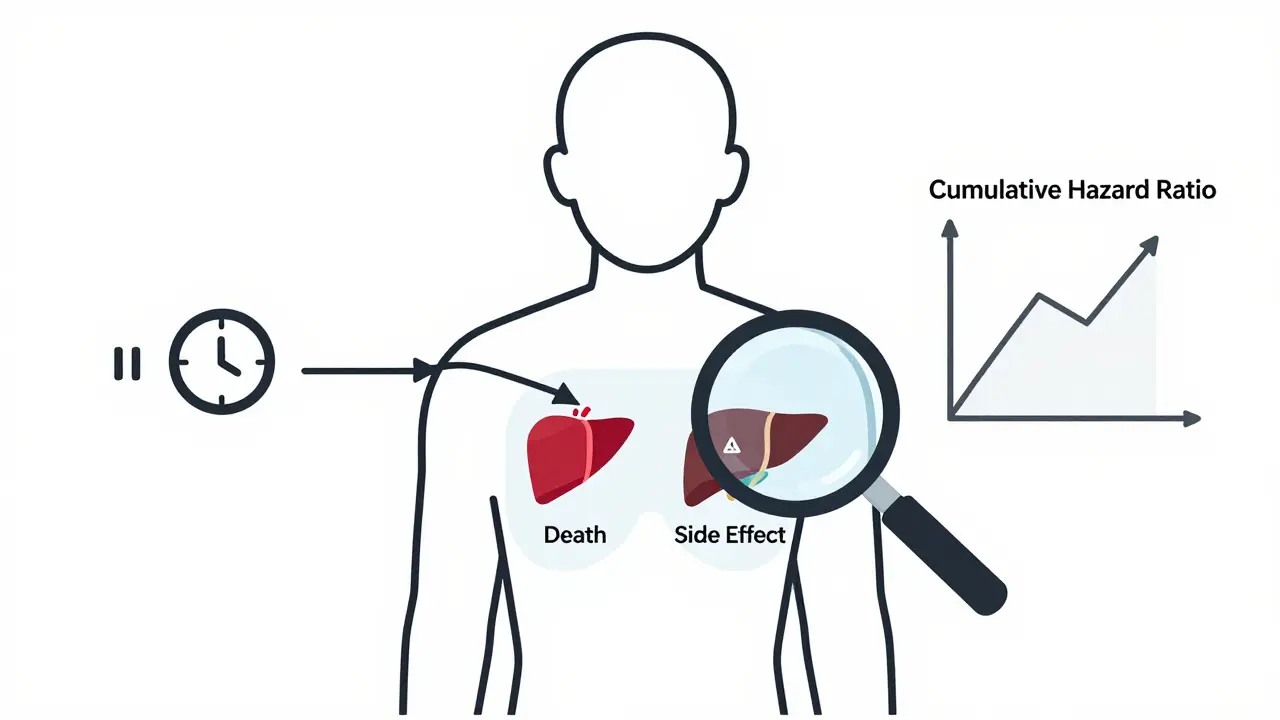
How Competing Risks Change Everything
Here’s where things get even trickier. In cancer trials or elderly populations, patients often die before they can experience a side effect. If someone dies from heart failure after 6 months, you never see if they’d have developed kidney damage at month 12.Traditional methods like the Kaplan-Meier estimator-commonly used in survival analysis-treat death as a "censored" event. But that’s wrong. Death isn’t just missing data-it’s a competing event. It removes the patient from the pool of people who could experience the adverse event.
A 2025 study in Frontiers in Applied Mathematics and Statistics showed that using Kaplan-Meier in these cases leads to inflated risk estimates. Instead, they recommend cumulative hazard ratio estimation, which breaks down risk into separate pathways: one for death, one for the adverse event. Simulations showed this method was 22% more accurate when competing events occurred in more than 15% of patients.
Real-World Challenges in Implementation
Switching from IR to EAIR sounds great-but it’s not easy. A 2024 PhUSE survey found that programming EAIR in SAS takes 3.2 times longer than IR. Median time: 14.7 hours vs. 4.5 hours. Common mistakes? Incorrect date handling (28% of cases), not accounting for treatment interruptions (19%), and inconsistent patient-year calculations (23%).Roche reported that 35% of their medical reviewers initially misread EAIR results. One reviewer thought a rate of 8.5 per 100 patient-years meant "8.5% of patients had the event." It doesn’t. It means "in every 100 people followed for one year, you’d expect 8.5 events." That’s not the same thing.
CDISC’s Oncology Therapeutic Area Guide (v3.0, 2023) now requires both IR and EAIR for serious adverse events. The FDA’s Biostatistics Review Template now includes checklists asking: "How was exposure time calculated?" and "Was treatment interruption accounted for?"
The PhUSE team released open-source SAS macros for EAIR calculation in March 2023. They’ve been downloaded over 1,800 times. Users report an 83% drop in programming errors after using them. Still, adoption is slow. Only 47% of regulatory submissions in 2023 included exposure-adjusted metrics-up from 12% in 2020, but far from universal.
What You Need to Know Today
If you’re reading clinical trial data-whether you’re a doctor, researcher, or patient-you need to ask three questions:- Is the adverse event rate reported as a percentage (IR) or as events per 100 patient-years (EIR/EAIR)?
- What’s the average treatment duration in each group? If it’s more than 6 months, raw percentages are unreliable.
- Was exposure time adjusted for treatment interruptions? If not, the numbers may be misleading.
The global clinical trial safety software market hit $1.84 billion in 2023, growing at 22.7% yearly-mostly because regulators are demanding better methods. The ICH E9(R1) guidelines from 2020 already require that safety analyses consider exposure time. The FDA’s 2024 draft guidance on exposure-adjusted analysis is just codifying what smart statisticians have known for years.
By 2027, experts predict 92% of Phase 3 drug submissions will include EAIR alongside IR. The future of drug safety isn’t about counting people-it’s about counting time, events, and real-world behavior.
What’s the difference between incidence rate (IR) and exposure-adjusted incidence rate (EAIR)?
Incidence rate (IR) is the percentage of patients who experienced an adverse event, regardless of how long they were on the drug. Exposure-adjusted incidence rate (EAIR) counts the number of events per unit of exposure time (usually per 100 patient-years), and adjusts for treatment interruptions, restarts, and varying durations between groups. EAIR gives a more accurate picture of how often side effects occur over time.
Why does the FDA prefer EAIR now?
The FDA prefers EAIR because raw percentages can hide real safety risks when treatment durations differ between groups. For example, a drug given for two years might naturally cause more side effects than one given for three weeks-but IR would make them look equally risky. EAIR accounts for time and recurrence, giving regulators a clearer, fairer view of true safety profiles. In 2023, the FDA required EAIR in an sBLA submission, marking a major regulatory shift.
Can I trust a study that only reports IR?
Be cautious. If the study has varying treatment lengths-especially over six months-IR alone is misleading. Always check whether exposure time was considered. A 2010 analysis showed IR can underestimate true event rates by up to 37% in trials with heterogeneous durations. Look for EAIR or EIR in safety tables. If it’s not there, the safety data may be incomplete.
What’s patient-year and how is it calculated?
A patient-year is one person exposed to a drug for one full year (365.25 days). To calculate it, you take the time between first and last dose for each patient, subtract any gaps where treatment was stopped, and divide by 365.25. For example: a patient on treatment for 18 months with a 2-week break = 1.5 years minus 0.04 years = 1.46 patient-years. Tools like JMP Clinical and SAS automate this using trial dates.
Do all regulatory agencies require EAIR?
No. The FDA is pushing hard for EAIR and now requires it in certain submissions. The European Medicines Agency (EMA) accepts both IR and EAIR but demands justification for the choice. The International Council for Harmonisation (ICH) E9(R1) guidelines require that safety analyses account for exposure time, but don’t mandate one specific method. Still, the industry trend is clearly moving toward EAIR as the gold standard.
What Comes Next
The FDA’s Sentinel Initiative is testing machine learning models that use EAIR data to automatically flag safety signals. Early tests showed a 38% improvement in detecting problems before they become widespread. Meanwhile, the PhUSE team is preparing an R-based reference implementation for EAIR, due in early 2025. As these tools become standard, the days of reporting simple percentages will be over.If you’re reviewing a clinical trial, don’t just look at the headline number. Ask: How much time did people spend on the drug? Were interruptions accounted for? Was recurrence measured? Because in drug safety, time isn’t just a factor-it’s everything.




14 Comments
So let me get this straight-FDA is forcing EAIR now? LOL. They’re just trying to bury the truth that Big Pharma’s been hiding side effects for decades. You think this is about science? Nah. It’s about control. They want us to trust their numbers while the real data gets buried in fancy math. I’ve seen it before-every time they change the metric, it’s because the old one exposed too much. This is a smoke screen. Watch for the next ‘mandatory’ change in 2026. It’ll be even more opaque. They don’t want you to understand. They want you to obey.
And don’t even get me started on how the ‘open-source SAS macros’ were released right after a major drug recall. Coincidence? I think not.
Actually the EAIR method is not new it has been used in epidemiology since the 1980s for infectious disease studies and its adoption in clinical trials is long overdue. The problem with IR is that it assumes uniform exposure which is statistically invalid when treatment durations vary. The 2010 Amgen analysis you mentioned is correct but understates the issue-some studies showed up to 45% underestimation when comparing 30-day vs 730-day cohorts. The FDA requirement is minimal. We should be using time-to-event models with competing risks as standard. Also patient-years must be calculated using actual dosing dates not approximations. Many still use average duration which introduces bias. Use TRTSDTM and TRTEDTM fields always.
Let’s be real-Americans are the only ones who care about this stuff. Europe still uses IR because they’re too lazy to update their systems. The FDA didn’t ‘push’ EAIR because it’s science-they pushed it because American litigators were getting too good at using IR flaws in lawsuits. You think this is about patient safety? Nah. It’s about liability. Pharma companies have been getting burned for years because some junior analyst counted patients instead of time. Now they’re forcing everyone to use EAIR so they can’t be sued for ‘misrepresenting risk.’ It’s not a breakthrough. It’s a legal shield.
I’ve been reading through this and I just want to say how deeply thoughtful this is. It’s so easy to get caught up in percentages and think you’re seeing the full picture, but time is such a silent variable. I work with elderly patients in hospice care, and I’ve seen so many cases where someone would get a side effect after 14 months, but since the trial only tracked 6 months, it was never counted. That’s not just a statistical error-that’s a moral one. When we reduce human experience to numbers without context, we fail people. The EAIR method doesn’t just fix the math-it restores dignity to how we measure harm. I hope more researchers take this seriously. Time isn’t just a unit. It’s life.
Also, the part about competing risks? That’s huge. Death isn’t censoring-it’s a different path. We need to stop pretending it’s just missing data. That’s not science. That’s denial.
The shift from IR to EAIR represents a necessary evolution in pharmacovigilance methodology. While IR provides a simple metric for initial risk assessment, its inability to account for differential exposure durations introduces systematic bias, particularly in chronic disease trials. EAIR, by normalizing event frequency per unit of exposure time, enables more accurate intercohort comparisons. Furthermore, the incorporation of treatment interruption windows aligns with real-world adherence patterns, improving external validity. Regulatory harmonization under ICH E9(R1) is a positive development, though implementation challenges remain, particularly in legacy datasets with incomplete TRTSDTM/TRTEDTM records. The PhUSE macros represent a pragmatic solution, though standardization of exposure calculation logic across sponsors remains inconsistent.
Why are we even talking about this? Nobody cares. You think patients read the safety section of a clinical trial? No. They read the headline: "75% effective!" and "only 10% had side effects." That’s it. The FDA doesn’t care about EAIR-they care about getting drugs approved faster. This whole thing is just a way for statisticians to feel important. I’ve been in pharma for 20 years. Nobody uses EAIR unless they’re forced to. And even then, they bury it in appendix D. The real problem? Patients don’t know what patient-years even mean. So why are we pretending this changes anything? It’s all theater.
How quaint. You’ve managed to turn a simple concept-time matters-into a 2000-word lecture. EAIR is not revolutionary. It’s basic epidemiology. The fact that this is even controversial reveals how deeply broken clinical trial design has become. The real scandal isn’t that IR is flawed-it’s that regulators didn’t catch this decades ago. And now we’re celebrating a 2023 mandate like it’s the moon landing? Please. The industry has been gaming IR since the 1990s. You think the 12% of safety signals revealed by EAIR are new? They’ve been there all along. They just got buried under lazy math. The only thing new here is the PR spin. And the fact that you’re all acting like this is a breakthrough? That’s the real tragedy.
Did you know the FDA’s EAIR requirement was pushed by a former Pfizer exec who now works at the agency? Yeah. That’s not a conflict of interest-that’s a revolving door. And the open-source SAS macros? They were written by a guy who used to work for Merck. Coincidence? I don’t believe in coincidences. This isn’t science. It’s a corporate reshuffle disguised as regulation. They’re not fixing safety-they’re locking out smaller companies who can’t afford the new software. The PhUSE macros? Downloaded 1800 times? That’s less than 1% of the industry. Most small CROs still use Excel. And they’re being forced to comply with EAIR? Good luck. This isn’t progress. It’s consolidation.
This made me pause and reflect. I’ve been a nurse for 15 years, and I’ve seen patients stop medications because they heard "15% had nausea"-but never knew how long they were on it. If someone took it for 3 weeks, 15% is terrifying. If they took it for 2 years? That’s normal. We don’t tell patients this. We just give them numbers. And they panic. Or worse-they don’t care. I wish more of this clarity made it to the patient-facing materials. The EAIR isn’t just for regulators-it’s for people trying to decide if they can live with the side effects. We owe them better. Also, thank you for mentioning competing risks. That’s something we never talk about. Death isn’t a footnote. It’s part of the story.
Good breakdown. Just wanted to add that the biggest hurdle isn’t the math-it’s communication. I’ve seen so many clinicians misinterpret EAIR as "8.5% of patients had the event" instead of "8.5 events per 100 patient-years." That’s a fundamental misunderstanding. We need training. Not just for reviewers, but for prescribers. A simple footnote on every safety table could help: "This is events per 100 patient-years, not percentage of patients." Small thing. Big impact. Also, kudos to PhUSE for the macros. They’re saving lives one line of code at a time.
Why is no one talking about the fact that EAIR still ignores individual variability? Two people can have the same exposure time, but one has a genetic mutation that makes them 5x more likely to have liver toxicity. EAIR treats them as identical. That’s not precision-that’s false precision. We’re replacing one flawed metric with another that’s just more complicated. We need personalized exposure modeling. Not just patient-years-patient-genome-years. Until then, we’re just rearranging deck chairs on the Titanic. And don’t get me started on how the FDA’s checklist ignores drug interactions. EAIR doesn’t care if you’re on warfarin. It just counts. That’s not safety. That’s negligence dressed up in statistics.
As someone from India who’s worked on global trials, I’ve seen firsthand how IR misleads in developing countries. In rural areas, patients often miss doses or stop early due to cost or distance. But IR still counts them as "exposed." EAIR fixes that. It’s not perfect, but it’s honest. I’m glad the FDA is pushing this. It’s not about nationalism-it’s about fairness. Everyone deserves accurate safety data, no matter where they live. Also, the 83% drop in errors with PhUSE macros? That’s huge. We need more open-source tools like this. Not just for big pharma, but for small clinics in Bihar or Nairobi. Science should be shared, not locked behind paywalls.
EAIR is still insufficient. You’re missing the concept of hazard function modulation. The assumption of constant hazard over time is invalid for most chronic therapies. The true metric should be the time-varying hazard rate, modeled via Cox regression with time-dependent covariates. EAIR is a crude approximation. The FDA’s mandate is a step, but not the destination. We need to move to individualized risk trajectories, not population-level aggregates. Also, the 2025 Frontiers study on cumulative hazard ratios? That’s the future. EAIR is 1990s thinking. We’re still in the stone age of safety analytics.
They say EAIR is more accurate? Please. The real reason they switched is because patients were suing over "15% nausea" when they were on the drug for 18 months. Now they can say "only 7.5 per 100 patient-years" and pretend it’s safer. It’s the same data. Just dressed up in jargon. I’ve reviewed dozens of submissions. The numbers never change. Only the words do. This isn’t science. It’s spin. And the FDA knows it. They just need a scapegoat for when things go wrong. EAIR gives them cover. That’s all.